-
Courses

Courses
Choosing a course is one of the most important decisions you'll ever make! View our courses and see what our students and lecturers have to say about the courses you are interested in at the links below.
-
University Life

University Life
Each year more than 4,000 choose University of Galway as their University of choice. Find out what life at University of Galway is all about here.
-
About University of Galway

About University of Galway
Since 1845, University of Galway has been sharing the highest quality teaching and research with Ireland and the world. Find out what makes our University so special – from our distinguished history to the latest news and campus developments.
-
Colleges & Schools

Colleges & Schools
University of Galway has earned international recognition as a research-led university with a commitment to top quality teaching across a range of key areas of expertise.
-
Research & Innovation

Research & Innovation
University of Galway’s vibrant research community take on some of the most pressing challenges of our times.
-
Business & Industry

Guiding Breakthrough Research at University of Galway
We explore and facilitate commercial opportunities for the research community at University of Galway, as well as facilitating industry partnership.
-
Alumni & Friends

Alumni & Friends
There are 128,000 University of Galway alumni worldwide. Stay connected to your alumni community! Join our social networks and update your details online.
-
Community Engagement

Community Engagement
At University of Galway, we believe that the best learning takes place when you apply what you learn in a real world context. That's why many of our courses include work placements or community projects.








Fleming Group
Mechanistic computational modeling of brain metabolism
Extensive manual curation of the biochemical literature, together with transcriptomic and proteomic data are combined with novel algorithms to reconstruct metabolic models specific to certain neuronal sub-types, e.g., dopaminergic neurons. Constraint-based computational modeling is used to model metabolism at genome-scale. Iterative rounds of reconstruction, model prediction and reconciliation with existing experimental data is used to develop a computational model which is formal synthesis of current knowledge on neuronal metabolic function. The accuracy of the neuronal metabolic model is tested by comparison with independent experimental data. Computational models of neuronal metabolism are used as an aid to interpret experimental data, to optimally design in vitro experiments with stem-cell derived neurons, to understand the aetiopathogenesis of neurodegenerative disease, e.g., Parkinson’s disease, and to develop new approaches for early diagnosis and treatment of disease.
Automated microfluidic cell culture
Automated microfluidic cell culture has various advantages over manual, macroscopic cell culture, including increased precision, ease of conducting perfusion culture, direct coupling to downstream analysis systems and the potential for real time on-chip analyses. In collaboration with Prof. Thomas Hankemeier, Leiden University, we are working toward the establishment of automated microfluidic cell culture of stem-cell derived cultures, coupled to analytical chemical measurement of metabolite concentrations. This, quantitative data is subsequently integrated with corresponding mechanistic models of neuronal metabolism.
Algorithms and software for mechanistic modeling of biochemical networks
In experimental systems biology, the majority of high throughput experimental data is of molecular abundance and the minority is of reaction rates. We seek a modeling framework flexible enough to integrate experimental data on both rates and abundance. Established genome-scale modeling methods do not explicitly represent the abundance of each molecule. Without explicitly representing abundance, the incorporation of experimental constraints from measurement of molecular abundance is always an approximation. Such approximation may be useful in the short to medium term, but ultimately we seek a computational model of biochemical reaction networks to explicitly represent the abundance of each molecule and the rate of each reaction. We are developing a novel approach to model stationary state reaction kinetics for large systems of reactions based on nonlinear continuous optimization algorithms. These variational kinetic models aim to preserve the computational tractability associated with numerical optimization and add the biochemical realism typical of kinetic models. Our general approach is to focus on the development of biochemically tailored polynomial-time optimization algorithms with guaranteed convergence properties. This effort requires the development and application of mathematical concepts at the intersection of Variational Analysis, Continuous Nonlinear Optimization, Generalised Convexity and Numerical Analysis. In tandem with mathematical algorithm development, we prototype numerical analysis software then test its performance when applied to a set of low, medium and high dimensional biochemically relevant modeling problems.
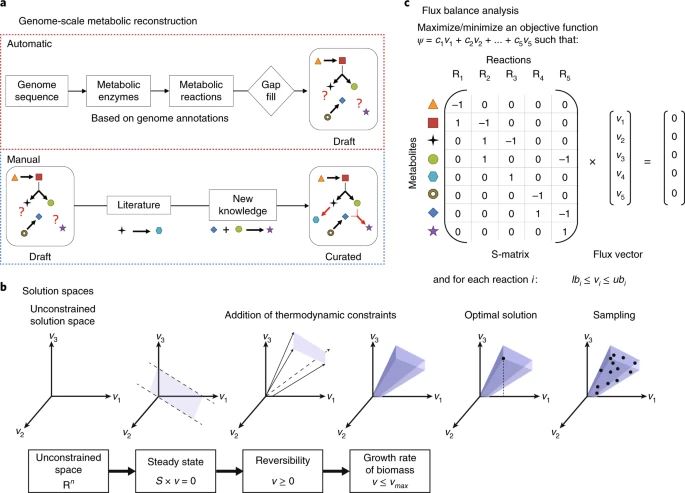
a, A genome-scale metabolic reconstruction is a structured knowledge base that abstracts pertinent information on the biochemical transformations taking place within a chosen biochemical system, e.g., the human gut microbiome. Genome-scale metabolic reconstructions are built in two steps. First, a draft metabolic reconstruction based on genome annotations is generated using one of several platforms. Second, the draft reconstruction is refined on the basis of known experimental and biochemical data from the literature6. Novel experiments can be performed on the organism and the reconstruction can be refined accordingly. b, A phenotypically feasible solution space is defined by specifying certain assumptions, e.g., a steady-state assumption, and then converting the reconstruction into a computational model that eliminates physicochemically or biochemically infeasible network states. Various methods are used to interrogate the solution space. For example, optimization for a biologically motivated objective function (e.g., biomass production) identifies a single optimal flux vector (v), whereas uniform sampling provides an unbiased characterization via flux vectors uniformly distributed in the solution space. c, Flux balance analysis is an optimization method that maximizes a linear objective function, ψ(v) = cTv, formed by multiplying each reaction flux vj by a predetermined coefficient, cj, subject to a steady-state assumption, Sv = 0, as well as lower and upper bounds on each reaction flux (lbj and ubj, respectively).
Please see here for research publications.
Funding
- European Union Horizon 2020








